")
 (ES)")
PANGENOMA
Anvio tiene un flujo de trabajo pangenómico que consta de varios pasos:
El directorio contiene bases de datos anvi'o contigs, un archivo de genomas externo y un archivo de datos delimitado por TAB que contiene información adicional para cada genoma (que es opcional, pero verá más adelante por qué es muy útil). Puede generar un almacenamiento de genomas como se describe en esta sección de la siguiente manera:
1. Generar una base de datos genómica Anvio está almacena secuencias de ADN y aminoácidos así como la anotación funcional de cada gen a partir de archivos fasta.
2. Calcular el pangenóma a partir de una base de datos que identifica los grupos de genes, está base de datos se realiza delimitando por medio de parámetros las similitudes de cada secuencia de aminoácidos en cada genoma contra cualquier otra secuencia de aminoácidos usando blastp. También, elimina los hits con menos puntajes entre las lecturas e identifica grupos de genes, calcula la aparición de grupos de genes entre genomas y el número total de genes que contienen, realiza análisis de agrupación jerárquica para grupos de genes según la distribución entre los genomas.
3. Muestra el pangenoma y visualiza la distribución de los grupos de genes a través de los genomas y de forma interactiva integra estos los grupos de genes en colecciones lógicas, también inspecciona la alineación de los genes en un grupo.
4. Calcular y visualizar puntuaciones promedio de identidad de nucleótidos entre sus genomas y más.
5. Finalmente, puede realizar un resumen de los datos encontrados en formato html.
Comando:
| anvi-gen-genomes-storage -e external-genomes.txt -o GENOMES.db |

Una vez que tenga listo el almacenamiento de sus genomas, puede usar el programa anvi-pan-genome para ejecutar el análisis pangenómico real. Cada parámetro después del --project-name es opcional (pero se alinea con la forma en que ejecutamos el pangenoma para nuestro estudio).
Además, con el flujo de trabajo pangenómico existe la opción de poder calcular y visualizar por medio de un mapa de calor el promedio de identidad de nucleótidos (ANI) entre los genomas que se estudian, el programa acepta archivos con formato fasta y el interpreta que cada fasta es un genomas. ANI se define como la identidad de nucleótidos media de pares de genes ortólogos compartidos entre dos genomas microbianos.
A continuación se muestra los comandos necesarios para poder extraer dicha información:
Comando para los Bacillus:
Una vez que tenga listo el almacenamiento de sus genomas, puede usar el programa anvi-pan-genome para ejecutar el análisis pangenómico real. Cada parámetro después del --project-name es opcional (pero se alinea con la forma en que ejecutamos el pangenoma para nuestro estudio).
Además, con el flujo de trabajo pangenómico existe la opción de poder calcular y visualizar por medio de un mapa de calor el promedio de identidad de nucleótidos (ANI) entre los genomas que se estudian, el programa acepta archivos con formato fasta y el interpreta que cada fasta es un genomas. ANI se define como la identidad de nucleótidos media de pares de genes ortólogos compartidos entre dos genomas microbianos.
A continuación se muestra los comandos necesarios para poder extraer dicha información:
Comando para los Bacillus:
|
anvi-pan-genome -g GENOMES.db --project-name "Bacillus_Pan" --output-dir Bacillus --num-threads 10 --minbit 0.5 --mcl-inflation 6 --use-ncbi-blast |
Comando para los Streptomyces:
|
anvi-pan-genome -g GENOMES.db --project-name "Streptomyces_Pan" --output-dir Streptomyces --num-threads 10 --minbit 0.5 --mcl-inflation 6 --use-ncbi-blast |
El directorio que ha descargado también contiene un archivo llamado "layer-optional-data.txt", que resume el clado al que pertenece cada genoma. Una vez que se calcula el pangenoma, podemos agregarlo a la base de datos de pan:
Comando para los Bacillus:
|
anvi-compute-genome-similarity --external-genomes external-genomes.txt \ --program pyANI \ --output-dir ANI \ --num-threads 10 \ --pan-db Bacillus/Bacillus_Pan-PAN.db |
Para poder visualizarlo en la pantalla interactiva deberá ingresar con el usuario reservando la dirección de IP, con el siguiente comando:
ssh -L 8080:127.0.0.1:8080 suusuario@kabré.cenat.ac.cr
|
anvi-display-pan -g GENOMES.db -p Bacillus/Bacillus_Pan-PAN.db |
Comando para los Streptomyces:
|
anvi-compute-genome-similarity --external-genomes external-genomes.txt \ --program pyANI \ --output-dir ANI \ --num-threads 10 \ --pan-db Bacillus/Bacillus_Pan-PAN.db |
De la misma forma en el caso de los Streptomyces deberá ingresar con el usuario reservando la dirección de IP, con el siguiente comando:
ssh -L 8080:127.0.0.1:8080 suusuario@kabré.cenat.ac.cr
| anvi-display-pan -g GENOMES.db -p Streptomyces/Streptomyces_Pan-PAN.db |
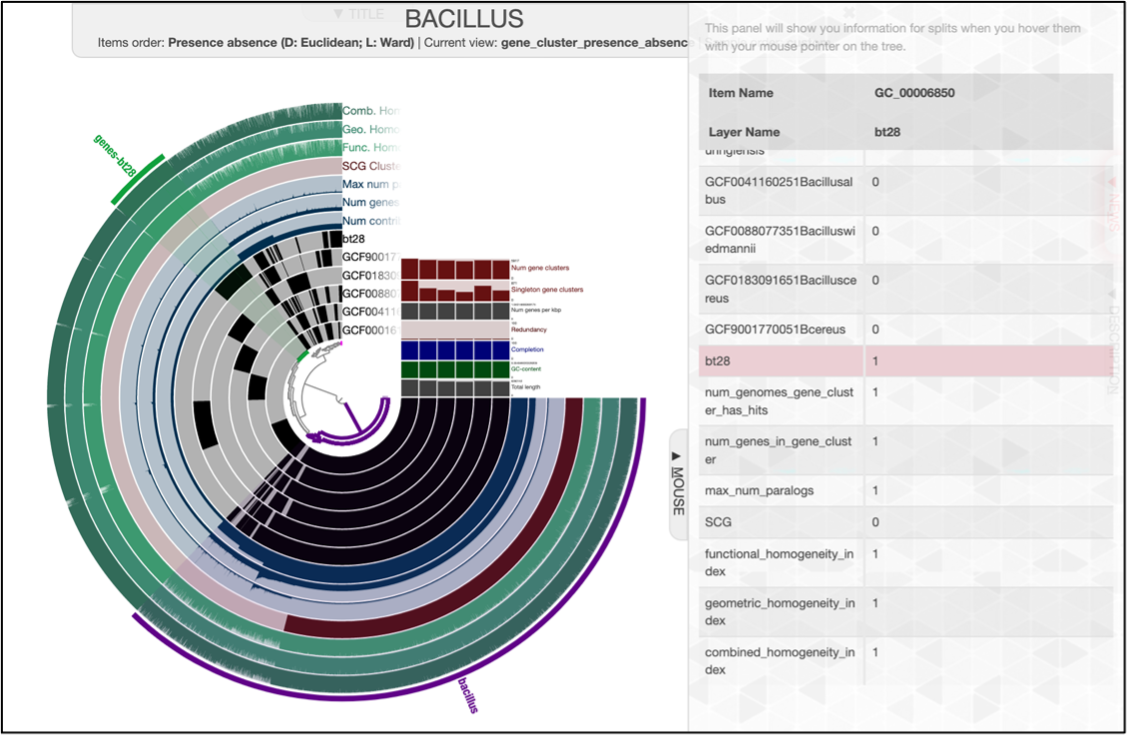
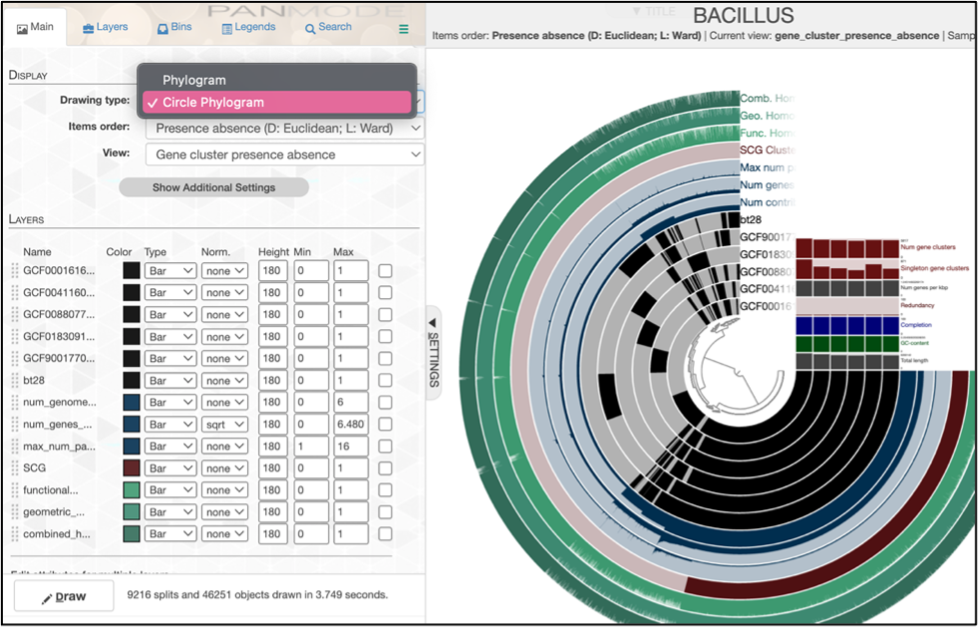
En este punto, al ingresar a su explorador con la ip 127.0.0.1:8080 o localhost, podrá observar al oprimir el botón “Draw” la imagen del dendograma con los genomas elegidos para realizar la pangenómica como se ve a continuación, además podrá elegir las configuraciones que le quiera poner a sus genomas como tamaño, color y forma del dendograma :
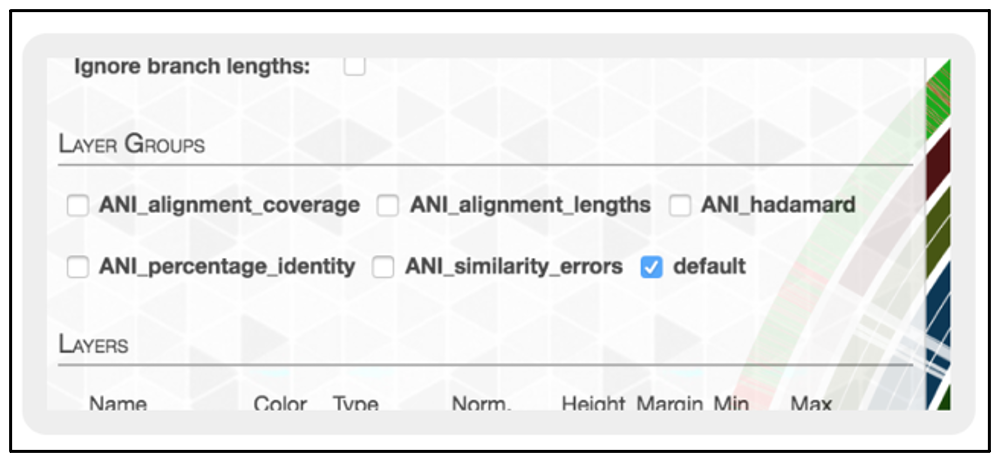
En la sección layers encontrará la opción para poder realizar las configuraciones ANI :
Una vez realizadas todas las configuraciones deseadas, deberá volver a oprimir “Draw” para poder observar los cambios realizados, como se muestra a continuación :
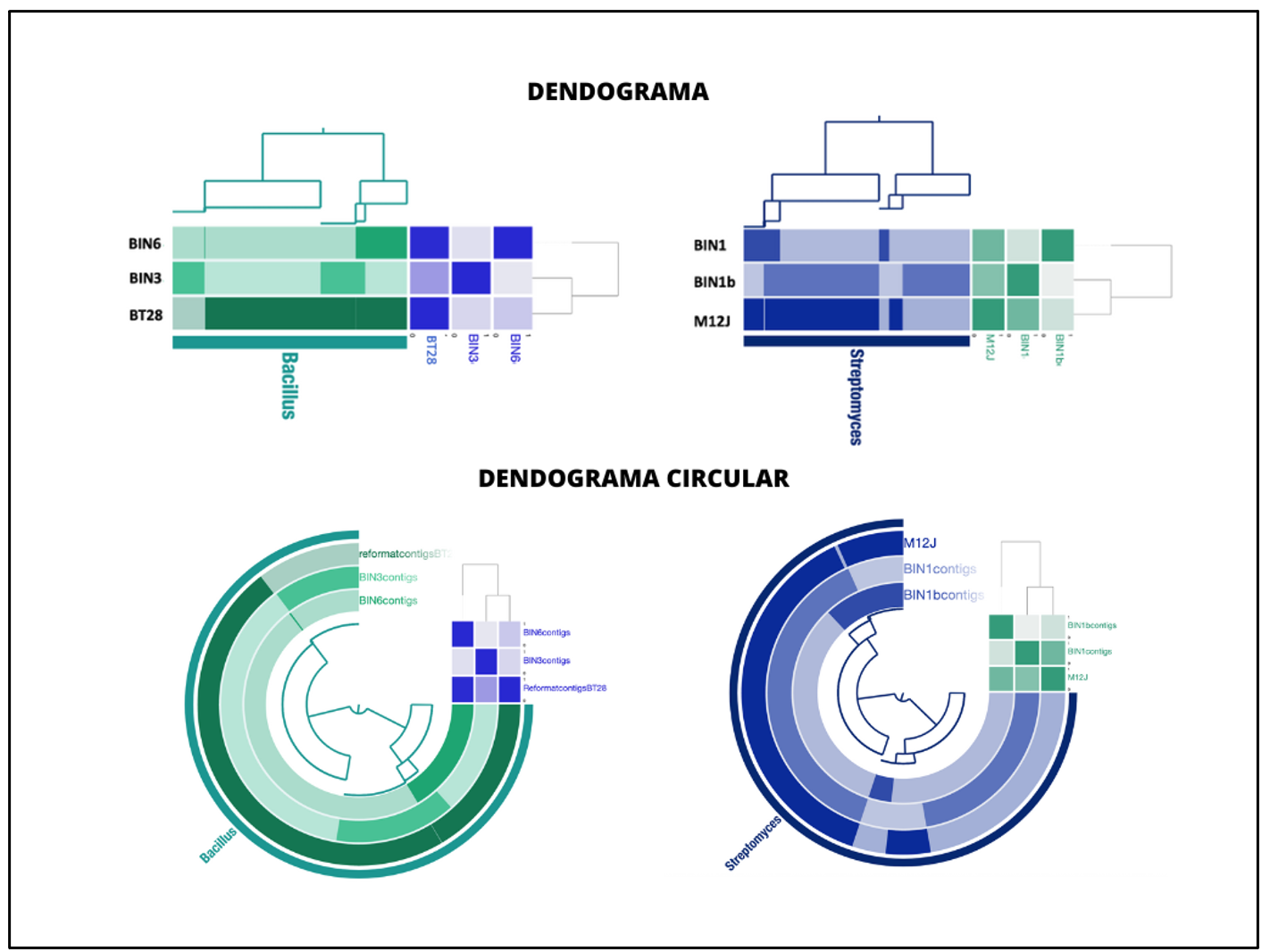
Los dendogramas mostrados anteriormente son creados a partir de los fasta extraidos del metagenoma artificial del estudio y los genomas individuales analizados, esto para mostrar los genes ortólogos que se están compartiendo entre ellos por medio del mapa de calor ANI y en el dendograma muestran la cantidad de grupo de genes estrechamente relacionadas entre genomas.
Finalmente, se repite el procedimiento anterior, esta vez incluyendo genomas de referencias extraídos de los resultados obtenidos de los genomas de referencia y los MAGs encontrados en el
metagenoma artificial con la herramienta GTDB-TK. Los resultados obtenidos fueron los que se muestran a continuación:
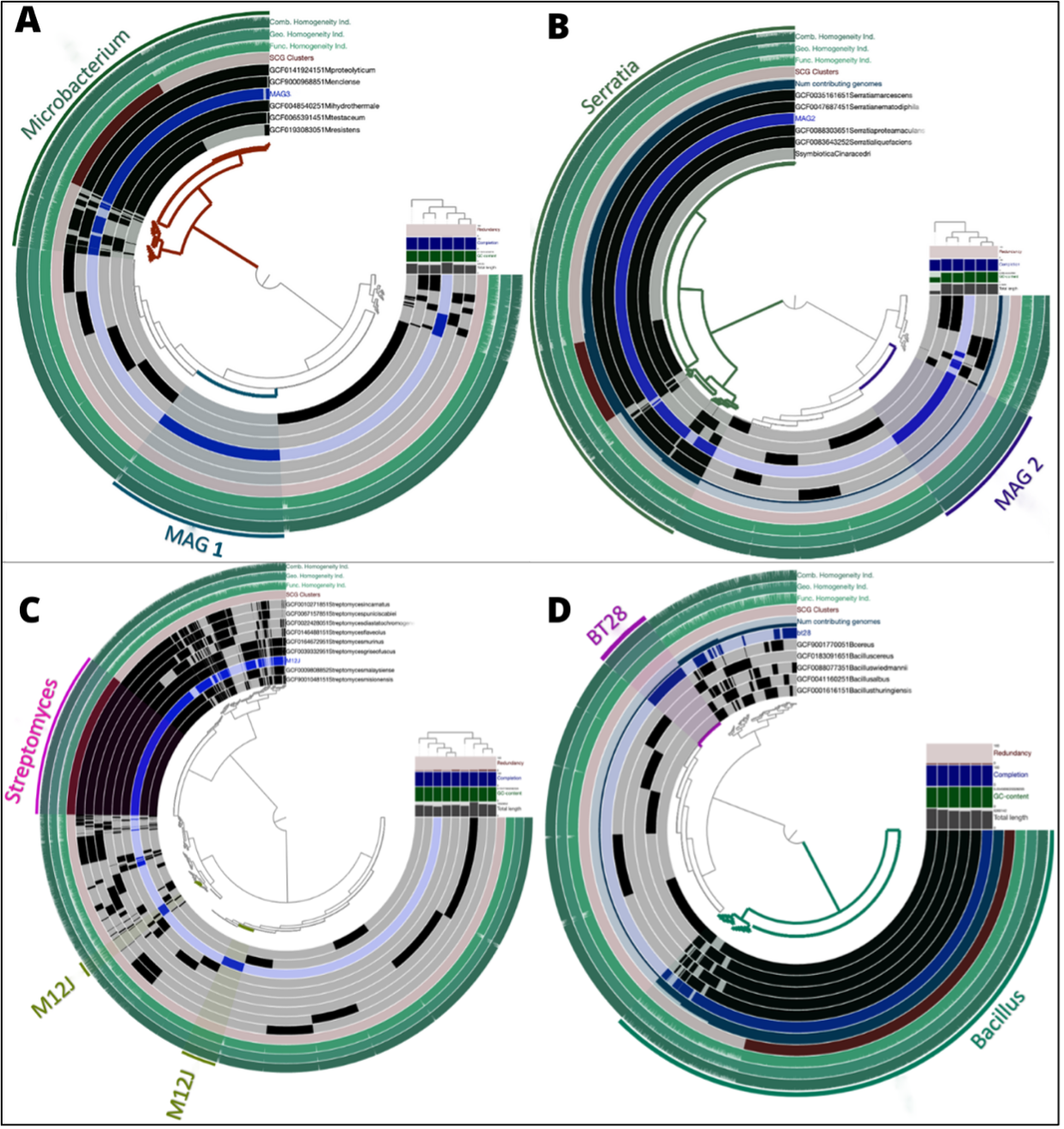
Las figuras muestra la gran similitud de los genes ortólogos encontrados entre los MAGS, genomas individuales y los genomas de referencia correspondientes A. Microbacterium, B. Serratia, C. Streptomyces y finalmente D. Bacillus. Además, Se marco en cada una de las figuras los segmentos de genes ortólogos únicos de los MAGS y genomas de referencia. En la pestaña “Mouse” muestra en detalle la sección marcada por el ratón del computador, mostrando en detalle la información del gen marcado.