Analisis de Anvio Metagenómico
PERFIL DE ANVIO
La base de datos contigs tiene toda la información necesario de los contigs del co-ensamblaje, ahora se va proporcionar información sobre cada una de las muestras a Anvio para que la herramienta pueda integrarlo todo. Cada muestra tendrá una base de datos de “perfil ” que mantendrá información de la muestra, las lecturas asignadas a cada contig y su ubicación.
| anvi-profile -i SAMPLE-01.bam -c contigs.d |

Desglose del código:
anvi-profile es el programa principal que estamos usando
● -i especifica el archivo bam de entrada específico de la muestra.
● -c nuestra base de datos contigs de entrada.
● -T especifica cuántos cpus usar.
MERGE
El siguiente paso es fusionar el flujo del trabajo de los perfiles de Anvio. El comando intentará crear múltiples agrupaciones de sus divisiones utilizando las configuraciones de agrupación predeterminada para los perfiles combinados si los hubiera. Anvio omitirá si tiene más de 20,000 divisiones, ya que la complejidad computacional de este proceso será menos factible con un número de divisiones.
| anvi-merge mapping/samples.bam-ANVIO_PROFILE/PROFILE.db mapping/samples2.bam-ANVIO_PROFILE/PROFILE.db -c reformat_contigs_fasta/contigs.db -o merged |
Desglose del código:
anvi-merge es el programa principal que estamos usando
● El primer argumento posicional (sin marca) específica las bases de datos de perfil de entrada específicas de la muestra. Para cada muestra, cuando ejecutamos anvi-profile anteriormente, la salida creó un subdirectorio de resultados. Al proporcionar el *comodín aquí seguido de a /PROFILE.db, los proporcionamos a todos como entradas para este comando.
● -o el directorio de salida.
● -c nuestra base de datos contigs.
VISUALIZACIÓN
La interfaz interactiva Anvio es una de las partes más sofisticadas, permite explorar los datos de manera intuitiva y logra ver de forma agradable los resultados de la agrupación de los datos. Es un entorno de visualización totalmente personalizable al que se puede manejar los datos de interés.
Se debe iniciar sesión un poco diferente para poder alojar el sitio web interactivo en un servidor local.
primero hay que entrar al cluster :
|
ssh -L 8080:127.0.0.1:Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo. |
Cargar los módulos normalmente y no cargar el comando:
(qsub -I -q debug)
Seguido, se ejecuta el siguiente comando:
|
anvi-interactive -p Merged/PROFILE.db -c contigs.db --server-only -I 127.0.0.1 -P 8080 |
Una vez se ejecuta el Anvi-interactive, puedes abrir el explorador web del computador y coloque en la barra del explorador http://localhost:8080 ó http://127.0.0.1:8080. Una vez cargada la página Anvio interactive haz click en “draw” y deberá aparecer el metagenoma que se está trabajando.
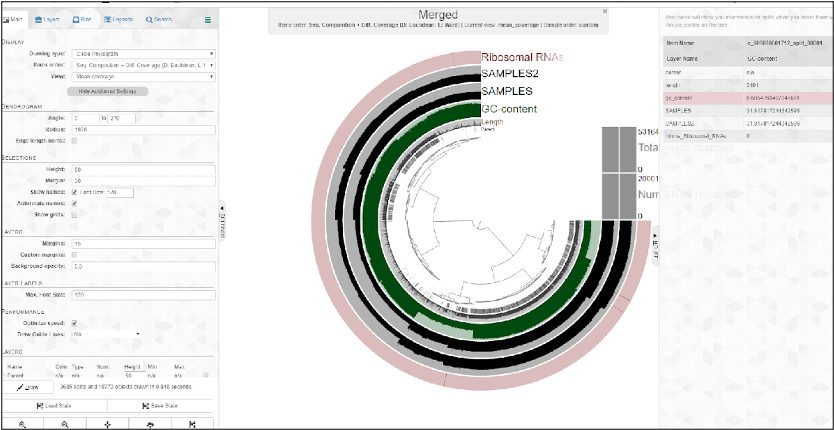
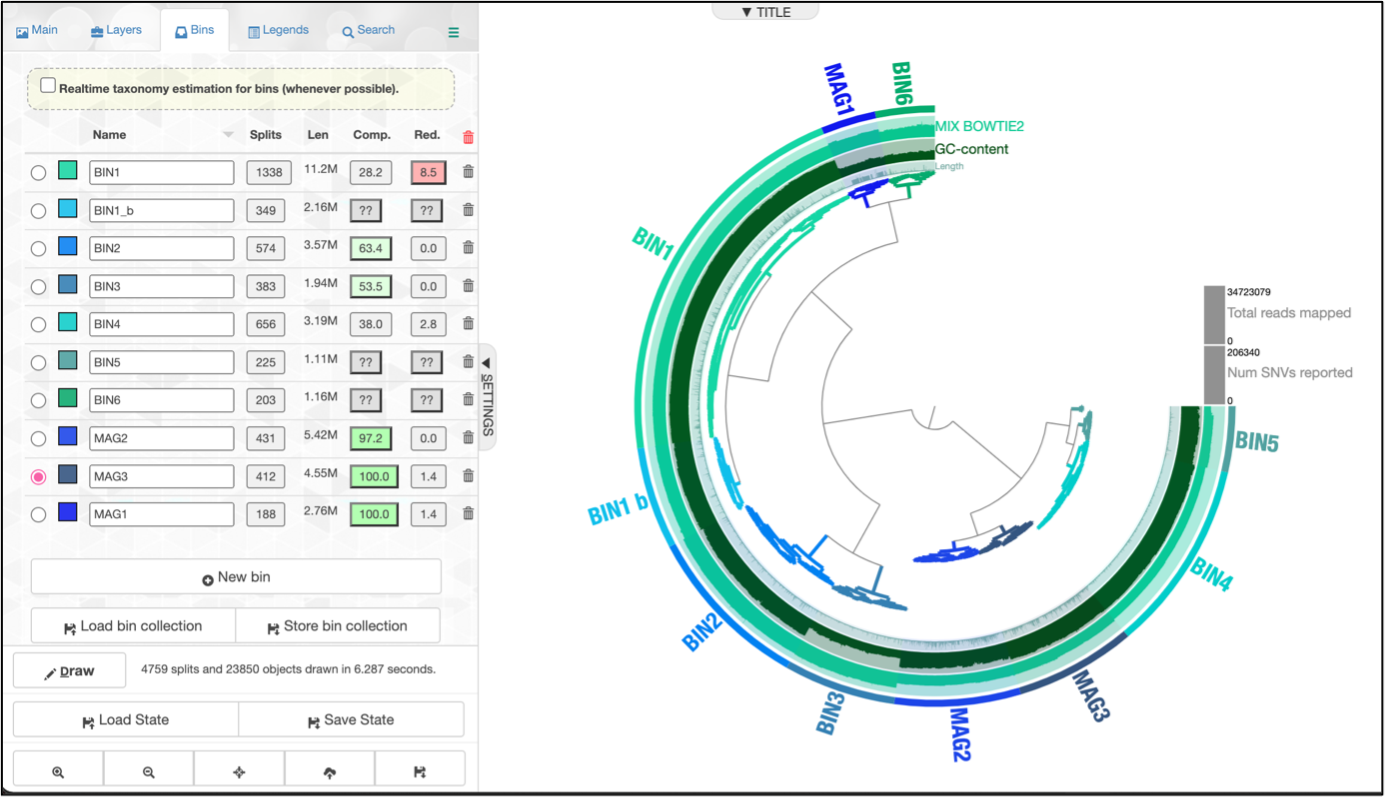
Una vez logre visualizar su muestra podemos ver cosas básicas como en el centro de la figura se muestra la agrupación jerárquica de los contigs del co-ensamblaje, es decir; cada rama del agrupamiento central es un contigs ó un fragmento de él. Se observa capas de información en la imagen como lo es su longitud, taxonomía, contenido de GC.
En la barra lateral derecha se muestra una opción llamada “mouse” donde se muestra un panel con toda la información el gen que nos interese visualizar en la imagine. Esto se logra moviendo el mouse del computador en las barras de la imagen de nuestra muestra.
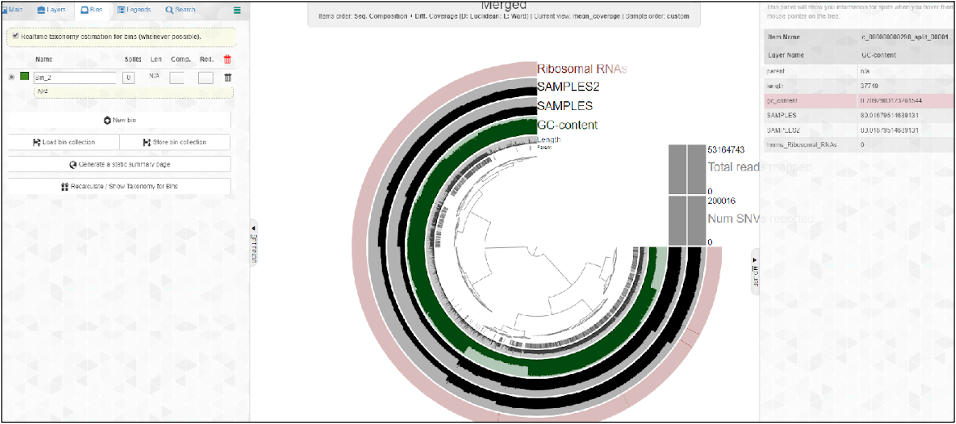
En la pestaña “bins” en el panel izquierdo permite crear agrupaciones manuales ya sean contigs metagenómicos, etiquetas, o cualquier tipo de información. La agrupación que se realice en esta interfaz se podrá crear,modificar y eliminar el “bin” seleccionado. También esta selección se puede realizar el almacenamiento de una nueva colección en el botón de “store bin collection”.
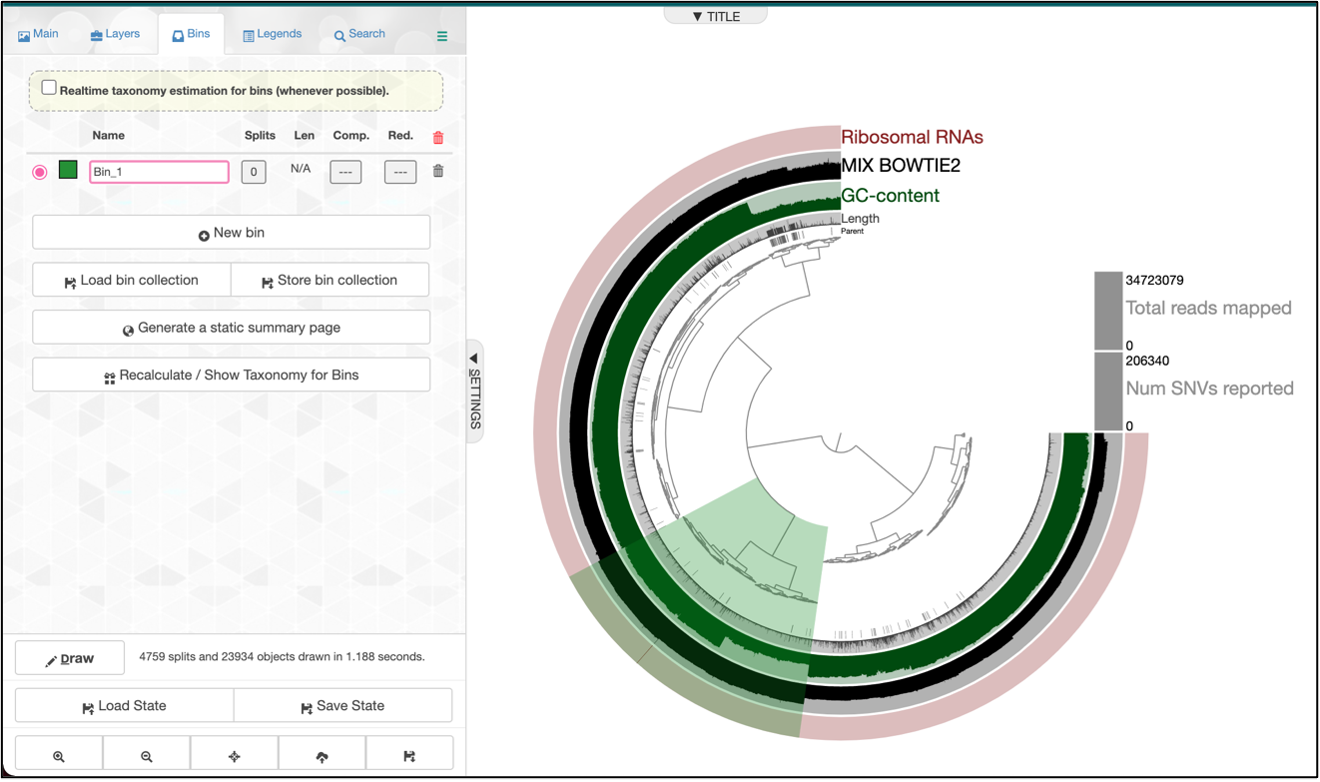
Cada vez que se crea el bin el panel muestra la longitud del bin creado con un porcentaje de completitud y de redundancia del bin creado. El porcentaje de completitud y redundancia proviene de los genes bacterianos de una sola copia que se analizó previamente con hmms.
Finalizada la selección de los bins y guardada la nueva colección, se puede generar un reporte con los datos relevantes de la colección. Se crea un documento html que se puede visualizar en el explorador. Una vez de utilizar el sistema interactivo se puede presionar control + C para cancelar la operación en el terminal.
El reporte generado de modo interactivo creó un directorio nuevo dentro de la carpeta Merge que se generó con anterioridad. Este nuevo reporte tiene información de la colección creada entre ellas los contigs creados en formato fasta. El dendograma generado se podría ver de la siguiente forma, aclarando que los nombres quedan a criterio del usuario:
FILOGENOMICA
Los análisis filogenómicos se pueden ejecutar con los archivos fasta, creados a partir de los genomas ensamblados con el metagenoma almacenados en la colección Anvio, también se podría generar un análisis filogenómico combinando los Mags creados en Anvio con algún otro recurso.
La filogenómica puede usar cualquier conjunto de genes para crear proteínas concatenadas, ya sea perfiles HMM creados en Anvio como perfiles HMM creados con otras herramientas.
De igual forma, los árboles filogenéticos creados se pueden visualizar de manera interactiva.
TRABAJANDO CON ARCHIVOS FASTA
Tenemos los archivos fasta creados en la sección anterior, lo primero que se va generar es una base de datos por cada uno de ellos en formato plano (adjunto en los archivos de ejemplo) con nombre external-genomes.txt.
Este archivo external-genomes.txt, lo tendremos incluído en el directorio donde se encuentran los archivos .fasta, esto para lograr realizar un archivo .db por cada contigs que queremos mostrar en el análisis.
El primero comando que se utilizará en esta sección lo incluiremos en un script para lograr hacerlos en un solo paso y no por cada archivo fasta que tenemos en el directorio.
|
for i in *fa do anvi-script-FASTA-to-contigs-db $i done |
Al finalizar, debería tener por cada archivo fasta un archivo con el mismo nombre pero con extensión .db.
Se utilizará un programa bastante poderoso que permite realizar muchos análisis con base de datos contigs, una colección de genomas externos o una base de datos de perfil.
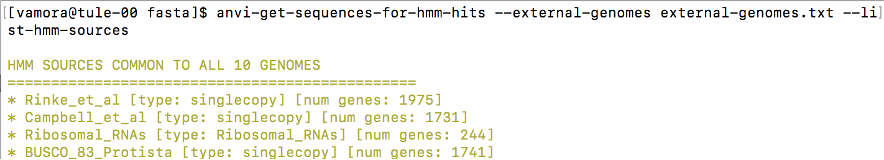
Para verificar cuales genes centrales de copia única se encuentran en la base de datos se utilizará el siguiente comando:
| anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt --list-hmm-sources |
De aquí en adelante se utilizará la colección Cambell et al. Sin embargo, se podría utilizar cualquier otra colección. Con el siguiente comando se podrán ver los genes que se identifican en la colección seleccionada:
| anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt --hmm-source Campbell_et_al --list-available-gene-names |

Ahora, se identifica cuál combinación de genes son las que se van a utilizar para el análisis filogenómico, inclusive todos los mostrados. Si no se declara como parámetro --gene-names, el programa utiliza todos los genes. El siguiente comando le dará las secuencias de aminoácidos concatenados en un archivo fasta.
| anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt -o concatenated-proteins.fa --hmm-source Campbell_et_al --return-best-hit --get-aa-sequences –concatenate |
Una vez se obtiene el archivo fasta se puede visualizar el archivo en la terminal utilizando el comando less seguido el nombre del archivo. Ahora sí, se podrá utilizar el programa que ayuda a obtener el árbol con formato newick de los genomas incluidos en el análisis.
| anvi-gen-phylogenomic-tree -f concatenated-proteins.fa -o phylogenomic-tree.txt |
Ahora, se tiene un archivo de salida con formato txt, el archivo contiene el árbol que muestra cómo se relacionan los genomas entre sí, y es momento de visualizarla en la interfaz Anvio. La primera vez que se ejecute el comando para visualizar el árbol en modo interactivo, Anvio genera automáticamente una base de datos de perfil vacía, si sigue este protocolo se hará de esa manera.
Primero hay que entrar al cluster :
| ssh -L 8080:127.0.0.1:8080 usuario@kabré.cenat.ac.cr |
Cargar los módulos normalmente y no cargar el comando. Seguido, se ejecuta el siguiente comando:
| anvi-interactive -p phylogenomic-profile.db -t phylogenomic-tree.txt --title "Phylogenomics" –manual --server-only -I 127.0.0.1 -P 8080 |
Una vez se ejecuta el Anvi-interactive, puedes abrir el explorador web del computador y coloque en la barra del explorador http://localhost:8080 ó http://127.0.0.1:8080.
De igual forma cuando se crearon los bins se mostrará una interfaz gráfica donde se muestra el botón “draw” y se mostrará el árbol creado de manera sencilla:
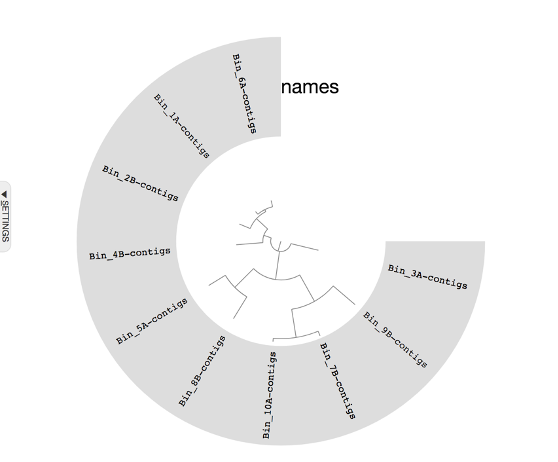
Después de darle un poco de formato con las herramientas interactivas que se muestran en las barras de herramientas Main podemos mejorar su visualización:
Como se puede ver, después de darle el formato deseado se logra visualizar de manera más agradable el árbol que realizamos. Se observa una distribución de los genomas aceptable, esta opción fue creada con la colección Anvio, pero como mencionamos anteriormente se puede generar un análisis filogenómico combinando los Mags creados en Anvio con algún otro recurso.
Agregamos archivos fasta externos del metagenoma estudiado, para verificar la correcta distribución de los datos en el árbol, y este fue el resultado:
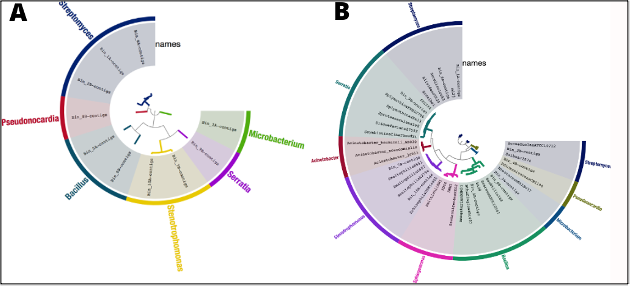
Se decidió agregar genomas de diferentes especies incluyendo los que se encontraron en el metagenoma de estudio, así como dos géneros independientes del estudio, esto para verificar el comportamiento de la herramienta. De manera muy positiva la herramienta logró identificar el género de cada genoma incluido en el estudio. Como se puede observar en la imagen.